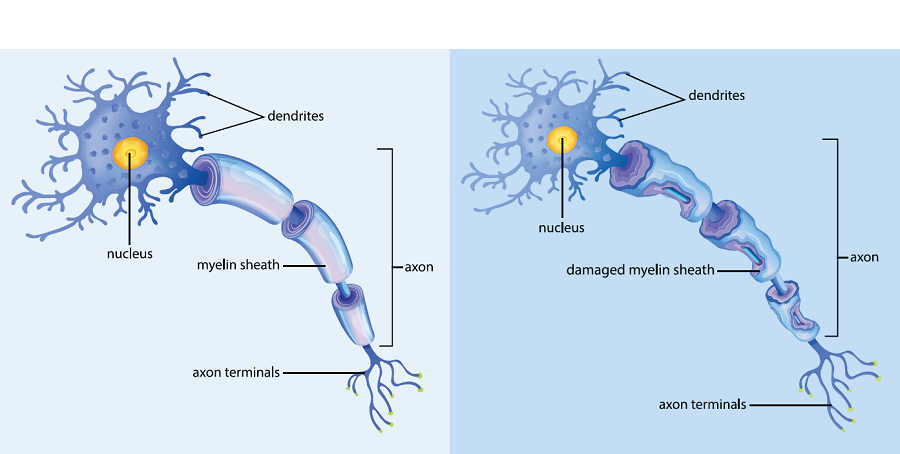
Molecular Pathogenesis of ADLD
Autosomal Dominant Leukodystrophy (ADLD) is a progressive neurodegenerative disorder driven by overexpression of the LMNB1 gene, caused either by a tandem genomic duplication of LMNB1 or by large upstream deletions. Both mechanisms appear to disrupt the regulatory elements that normally control LMNB1 expression in oligodendrocytes, though the exact details remain an active area of investigation among multiple research groups.
At the cellular level, Lamin B1 accumulation likely impacts both oligodendrocytes and astrocytes:
-
Oligodendrocyte Dysfunction: Lamin B1 accumulation thickens the nuclear lamina, increasing nuclear rigidity. It acts as an epigenetic modulator, increasing repressive chromatin marks that downregulate lipid synthesis genes (like Scd1 and Elovl7), starving cells of the lipids required to maintain myelin. ADLD also acts as a spliceopathy; increased Lamin B1 upregulates RAVER2, leading to aberrant alternative splicing of the PLP1 transcript.
-
Astrocyte Reactivity: Astrocytes are a primary pathological driver. Lamin B1 overexpression triggers astrocytic reactivity and drastically reduces secretion of leukemia inhibitory factor (LIF), dampening Jak/Stat3 and PI3K/Akt survival signaling in neighboring cells and depriving myelinating oligodendrocytes of essential functional support.
Preclinical Models and Biomarker Discovery
Therapeutic development utilizes the PLP-LMNB1Tg mouse model, which targets Lamin B1 overexpression specifically to oligodendrocytes and successfully recapitulates the late-onset, age-dependent demyelination, ataxia, and severe motor dysfunction seen in human patients. On the in vitro front, researchers utilize patient-derived iPSC neurons and high-content drug screening platforms to map disease networks and evaluate transcriptomic readouts.
A critical bottleneck remains the lack of validated, quantitative biomarkers. Efforts are underway to identify reliable fluid biomarkers (plasma miRNAs, CSF lactate) and develop computational MRI grading systems to reliably track disease progression during clinical trials.
Clinical Practice Guidelines (2025)
The first consensus clinical practice guidelines for ADLD were published in 2025. Key conclusions: whole-exome sequencing (WES) is not recommended as a first-tier diagnostic tool because it often misses structural variants. Diagnosis requires single-gene testing capable of high-resolution structural variant detection. The guidelines also established standardized baseline and longitudinal surveillance protocols utilizing brain and cervical spine MRIs, tilt table testing, and neuropsychometric assessments.
References
- 1. Padiath, Q.S. (2019). Autosomal Dominant Leukodystrophy: A Disease of the Nuclear Lamina. Frontiers in Cell and Developmental Biology. Frontiers ↗
- 2. Bartoletti-Stella et al. (2015). Messenger RNA processing is altered in autosomal dominant leukodystrophy. Human Molecular Genetics. HMG ↗
- 3. Ratti et al. (2023). Understanding the Ultra-Rare Disease ADLD: an Updated Review. Molecular Neurobiology. Mol Neurobiol ↗
Access full publications, grant opportunities, and institutional partnership details.
View Resources